








The main objective of the GENDEV team is to decipher the genetic causes of neurodevelopmental disorders through the conduct of integrative and translational projects.
The team comprises several clinicians who give access to a wide range of neurogenetic disorders and a broad recruitment of well characterized patients through National Reference Centres for rare diseases, as well as molecular geneticists/biologists and bioinformaticians.
The presence of these complementary skills allows to develop ambitious projects that aim at elucidating underlying pathophysiological mechanisms.
The GENDEV team has access to the platforms of the CRNL (imaging, histology, molecular biology), and SFR Biosciences and Santé Lyon-Est (imaging, flow cytometry, genomics, zebrafish animal facility, etc.)
The team consists of:
- 2 Inserm researchers: Marion DELOUS and Sylvie MAZOYER
- 2 tenured UCBL1 engineers: Alicia BESSON and Anne MEILLER
-1 engineer: Fanny DESURMONT
- 4 PhD student (UCBL1/Inserm): Silvestre CUINAT (3A), Cyril JOVANI (4A), Esma KAHOUL (1A), Alexia RABEC (1A)
- 2 clinicians/geneticists: Patrick EDERY and Audrey PUTOUX
and many undergraduate students Master, License, ESTBB...
- To identify rare disease genetic basis: cohorts, family-based studies, linkage analysis, array CGH, next-generation sequencing (panel of genes, exome, whole genome)
- To study gene function, effects of mutations and pathophysiological mechanisms: human cell models (patients' cells, and induced pluripotent stem cells (iPSC)) and animals (zebrafish), molecular and biochemical approaches for the analysis of gene expression (qRT-PCR, RNAseq, etc.) and protein (immunoprecipitation, western blot, proteome…), confocal imaging on fixed/living samples…
- We also use data (transcriptomes…) from human or zebrafish public databases.
The two main projects of the team concern distinct pathologies:
- a rare Mendelian disease, the Taybi-Linder syndrome (TALS for TAybi-Linder Syndrome, or MOPD1 for microcephalic osteodysplastic primordial dwarfism type 1),
- Williams-Beuren and 7q11.23 microduplication syndromes, which are mirror-image neurodevelopmental disorders, one with a loss and the other with a gain of the 7q11.23 region.
Taybi-Linder Syndrome
Principal investigators :
Marion DELOUS, for the studies of the cellular and molecular (splicing) pathophysiological mechanisms with human iPSC-derived models and zebrafish models
Sylvie MAZOYER, for the studies on minor splicing and molecular diagnosis of new patients
Team members : Alicia BESSON (IE), Silvestre CUINAT (PhD student), Fanny DESURMONT (IE), Cyril JOVANI (PhD student), Esma KAHOUL (PhD student) Anne MEILLER (IE), Alexia RABEC (PhD student)
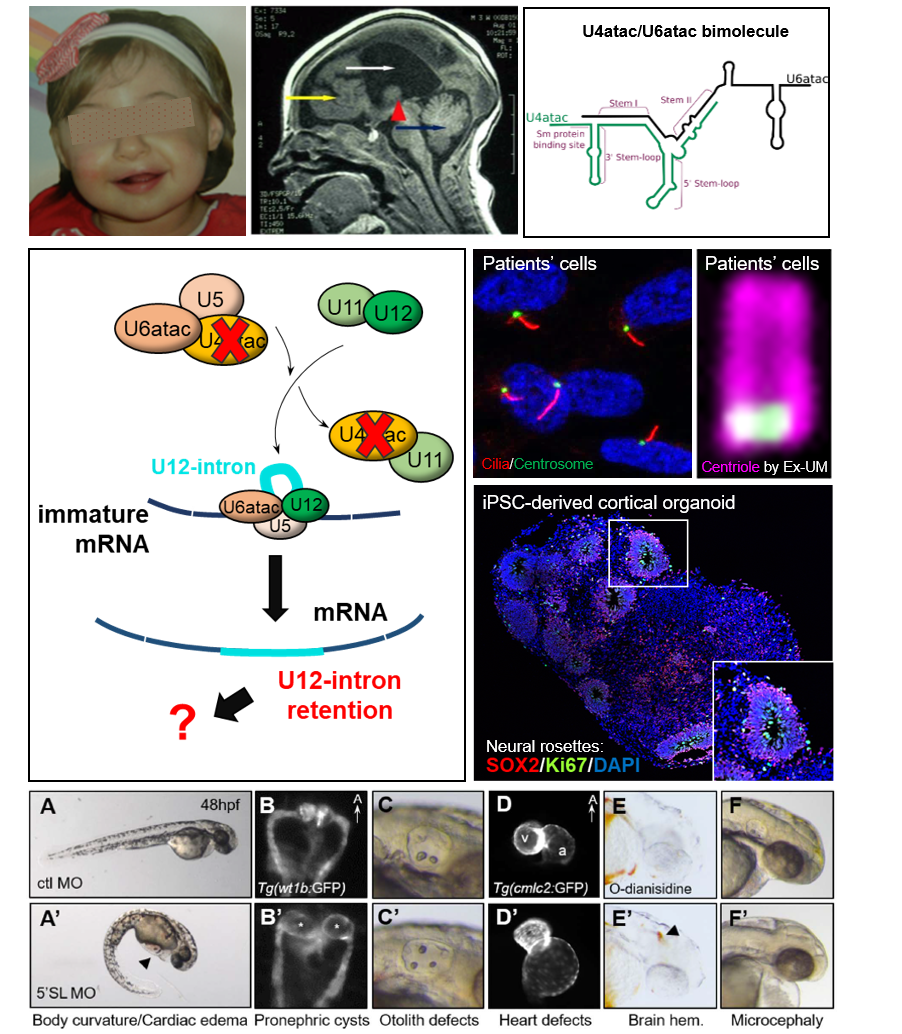
TALS is a very rare and severe autosomal recessive disorder characterized by syndromic brain malformations including severe neuronal migration defects, very short stature with bone anomalies, and early unexplained death (< 3 years old).
The causative gene, RNU4ATAC, was the first identified non-coding gene responsible for a monogenic disorder. U4atac snRNA is a component of the minor spliceosome involved in the removal of <1% of introns, the so-called U12-type introns (~850 introns located in 700 genes).
Recently, we discovered RNU4ATAC mutations in another pathology, Joubert syndrome (JBTS) which belongs to the class of ciliopathies. This discovery allowed us to make a link between minor splicing and the primary cilium, which we confirmed by various approaches (cellular models, zebrafish). Thus, we seek to understand why RNU4ATAC mutations lead 1- to ciliary anomalies, 2- to microcephaly and cerebral anomalies seen in TALS, 3- to different manifestations (TALS or JBTS). Our studies also aim to investigate the mechanisms controlling the splicing of U12-type introns, the role of the minor spliceosome remaining a great mystery to date.
To answer these questions, we use various models: patient-derived cells, iPS cells differentiated into neural progenitors/neurons and brain organoids, and the zebrafish model.
Fundings for TALS project
Contacts :
Marion Delous, for the studies of the cellular and molecular pathophysiological mechanisms using human iPSC-derived models and zebrafish models
Sylvie Mazoyer, for the studies on minor splicing and molecular diagnosis of new patients
7q11.23 chromosomal imbalances : Williams-Beuren syndrome and 7q11.23 microduplication syndrome
Principal investigator : Massimiliano ROSSI (PH)
Team member : Alicia BESSON (IE)
Williams-Beuren syndrome (SWB) is a rare genetic disease (incidence: 1/7500 -1/20000) caused by a 7q11.23 microdeletions. The phenotype includes a neurodevelopmental disorder characterized by social disinhibition, excessive talkativeness and mild to moderate intellectual disability, typical dysmorphic features and a vasculopathy (arterial stenosis, hypertension).
Microduplication of the same 7q11.23 chromosomal region causes a "mirror" syndrome (7q duplication syndrome/7qDS) characterized by a neurodevelopmental disorder of variable severity including autism spectrum disorders and selective mutism, mild dysmorphic features, and a vasculopathy (aortic dilatation).
The 7q critical region includes 26-28 genes including:
- GTF2I, BAZ1B, CLIP2, and EIF4H, which play a major role in the pathogenesis of SWB and 7qDS neurodevelopmental disorder;
- ELN, encoding elastin, whose haploinsufficiency is responsible for the vasculopathy of SWB. The pathogenesis of 7qDS vasculopathy is currently unknown.
Currently, there is no specific treatment for either neurodevelopmental disorders or vasculopathies observed in 7q11.23 chromosomal imbalances. The current research project aims to contribute to the characterization of the molecular and physiopathological mechanisms of 7q11.23 chromosomal imbalances and to the development of future therapeutic approaches.
This project includes a collaboration with the "High Definition Disease Modelling Lab: Stem Cell and Organoid Epigenetics, Centre for Neurogenomics" of the Human Technopole Institute in Milan, Italy and with Dr Aurore CURIE (Equipe Trajectoires, CRNL).
Funding of the project 7q11.23

Contact : Massimiliano Rossi