








L’objectif principal de l’équipe GENDEV est de décrypter les causes génétiques de maladies du neurodéveloppement à travers la conduite de projets intégratifs et translationnels.
L’équipe comprend plusieurs cliniciens qui donnent accès au recrutement d'un large éventail de patients avec des troubles neurogénétiques. Ceux-ci sont inclus et caractérisés par des centres nationaux de référence pour les maladies rares. L'équipe est aussi constituée de généticiens/biologistes moléculaires et de bioinformaticiens.
La présence de ces compétences complémentaires permet de développer des projets ambitieux visant à élucider les mécanismes pathophysiologiques sous-jacents.
L'équipe GENDEV a accès aux plateformes du CRNL (imagerie, histologie, biologie moléculaire), et des SFR Biosciences et Santé Lyon-Est (imagerie, cytométrie de flux, génomique, animalerie poisson zèbre,...)
L'équipe est constituée de :
- 2 chercheures Inserm : Marion DELOUS et Sylvie MAZOYER
- 2 ingénieures d'étude statutaires (UCBL1) : Alicia BESSON et Anne MEILLER
- 1 ingénieure (contractuelle) : Fanny DESURMONT
- 4 doctorants (UCBL1, Inserm) : Silvestre CUINAT (3A), Cyril JOVANI (4A), Esma KAHOUL (1A), Alexia RABEC (1A)
- 2 cliniciens/généticiens : Patrick EDERY et Audrey PUTOUX
et de nombreux stagiaires Master, Licence, ESTBB...
- Pour identifier les bases génétiques des maladies rares : cohortes, études familiales, analyses de liaison, analyse chromosomique sur puce à ADN (ou CGH-array), séquençage de nouvelle génération (panels de gènes, exomes, génomes entier)
- Pour étudier la fonction des gènes identifiés, les effets des mutations et les mécanismes physiopathologiques : modèles cellulaires humains (cellules de patients, et cellules souches pluripotentes induites (iPSC)) et animaux (poisson zèbre), approches moléculaires et biochimiques pour l’analyse de l’expression génique (qRT-PCR, RNAseq…) et protéique (immunoprécipitation, western blot, protéome…), imagerie confocale sur échantillons fixés/vivants...
- Nous utilisons également des données (transcriptomes…) obtenus à partir de lignées/tissus humains ou de poissons zèbres provenant de bases de données publiques.
Les deux principaux projets de l’équipe concernent des pathologies distinctes :
- une maladie mendélienne rare, le syndrome de Taybi-Linder (TALS pour TAybi-Linder Syndrome, ou MOPD1 pour nanisme primordial ostéodysplasique microcéphalique de type 1),
- les syndromes de Williams-Beuren et de microduplication 7q11.23, qui sont des troubles du neurodéveloppement en miroir, l'un avec une perte et l'autre avec un gain de la région 7q11.23.
Syndrome de Taybi-Linder
Principaux investigateurs :
Marion DELOUS (CR), pour l'étude des mécanismes physiopathologiques à l'aide des modèles humains dérivés d'iPSC et modèles poisson-zèbre
Sylvie MAZOYER (DR), pour l'étude de l'épissage mineur et de l'aide au diagnostic des nouveaux patients.
Membres d'équipe : Alicia BESSON (IE), Silvestre CUINAT (Doc.), Fanny DESURMONT (IE), Cyril JOVANI (Doc.), Esma KAHOUL (Doc.), Anne MEILLER (IE), Alexia RABEC (Doc.)
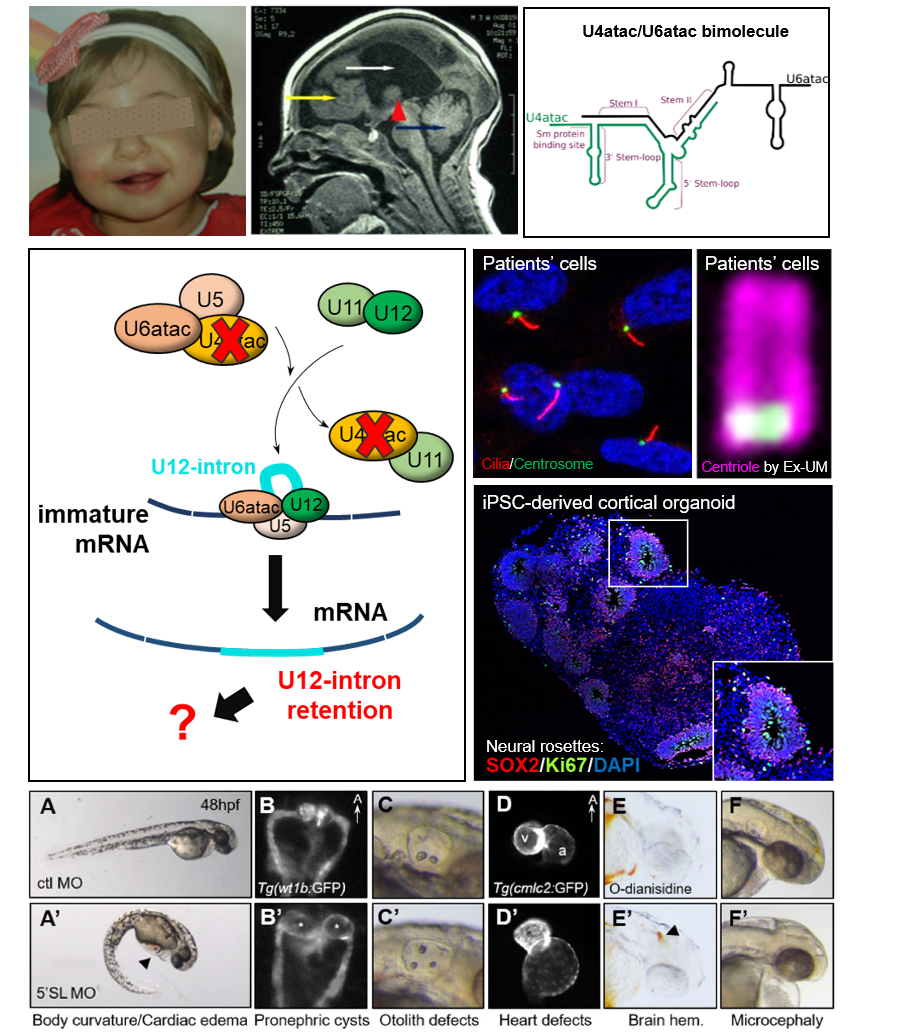
TALS est un syndrome autosomique récessif très rare et sévère caractérisé par une microcéphalie, des malformations cérébrales, notamment de graves anomalies de la migration neuronale, un nanisme, des anomalies osseuses et une mort précoce inexpliquée (<3 ans) dans les formes les plus sévères.
Le gène en cause, RNU4ATAC, a été le premier gène non codant identifié (par l'équipe) comme responsable d’une maladie monogénique. U4atac est un des ARNsn constituant le splicéosome mineur impliqué dans l’élimination de 1% des introns, les introns mineurs (ou de type U12), qui sont au nombre d’environ 935 situés dans 750 gènes.
Récemment, nous avons découvert des mutations de RNU4ATAC dans une autre pathologie, le syndrome de Joubert (JBTS) qui appartient à la classe des ciliopathies. Cette découverte nous a permis de faire un lien entre l'épissage mineur et le cil primaire, que nous avons confirmé par diverses approches (modèles cellulaires, poisson-zèbre). Ainsi, nous cherchons à comprendre pourquoi les mutations de RNU4ATAC conduisent 1- à des anomalies ciliaires, 2- à la microcéphalie et aux anomalies cérébrales vues dans TALS, 3- à des manifestations différentes (TALS ou JBTS). Nos études visent également à étudier les mécanismes contrôlant l’épissage des introns de type U12, le rôle du splicéosome mineur restant un grand mystère à ce jour.
Pour répondre à ces questions, nous utilisons divers modèles : des cellules dérivées de patients, des cellules iPS différenciées en progéniteurs neuronaux/neurones et organoïdes cérébraux, et le modèle poisson zèbre.
Financements du projet TALS
Contacts :
Marion Delous, pour l'étude des mécanismes physiopathologiques cellulaires et moléculaires, à l'aide des modèles humains dérivés d'iPSC et modèles poisson-zèbre
Sylvie Mazoyer, pour l'étude de l'épissage mineur et de l'aide au diagnostic des nouveaux patients
Déséquilibres chromosomiques 7q11.23 : Syndrome de Williams-Beuren et Syndrome de microduplication 7q11.23
Principal investigateur : Massimiliano ROSSI (PH)
Membres d'équipe : Alicia BESSON (IE)
Le syndrome de Williams-Beuren (SWB) est une maladie génétique rare (incidence : 1/7500 -1/20000) causée par une microdélétion 7q11.23. Le phénotype associe un trouble du neurodéveloppement caractérisé par une appétence sociale et une déficience intellectuelle légère à modérée, une dysmorphie et une vasculopathie (sténoses artérielles, hypertension).
La microduplication de la même région chromosomique 7q11.23 cause un syndrome « en miroir » (7q duplication syndrome/7qDS) caractérisé par un trouble du neurodéveloppement de sévérité variable incluant des troubles du spectre autistique et un mutisme sélectif, une dysmorphie discrète et une vasculopathie à type de dilatation aortique.
La région critique 7q inclut 26-28 gènes dont:
- GTF2I, BAZ1B, CLIP2 et EIF4H, qui ont un rôle majeur dans la pathogenèse du trouble du neurodéveloppement du SWB et du 7qDS;
- ELN, codant pour l’élastine, dont l’haploinsuffisance est responsable de la vasculopathie du SWB. La pathogenèse de la vasculopathie du 7qDS n’est actuellement pas connue.
Actuellement, il n’existe pas de traitement spécifique ni pour les troubles du neurodéveloppement ni pour la vasculopathie des déséquilibres chromosomiques 7q11.23. Le projet de recherche en cours vise à contribuer à la caractérisation des mécanismes moléculaires et physiopathologiques des déséquilibres chromosomiques 7q11.23 et au développement de futurs approches thérapeutiques.
Ce projet inclut une collaboration avec le Laboratoire "High Definition Disease Modelling Lab: Stem Cell and Organoid Epigenetics, Centre for Neurogenomics" de l’Institut Human Technopole à Milan, Italie et avec le Dr Aurore CURIE (Equipe Trajectoire, CRNL).
Financements du projet 7q11.23

Contact : Massimiliano Rossi